So you know about molecular biophysics and want to think about cells. How should you get started? Let’s take the first few steps.
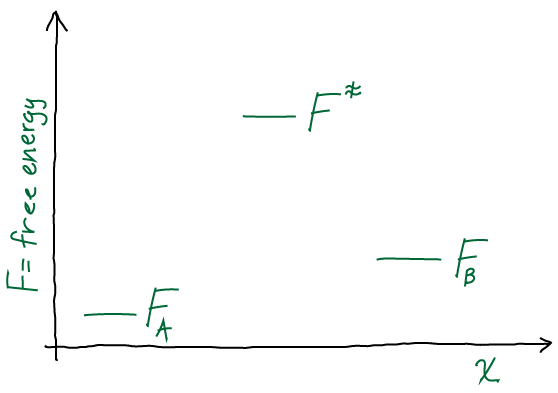
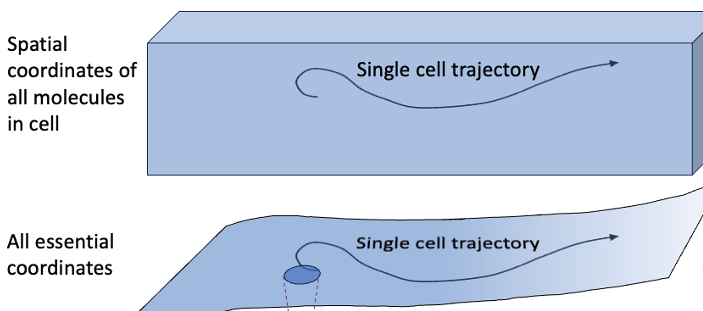
We’ll start with some concepts that should be familiar: coordinates and equilibrium vs. nonequilibrium behavior. We can frame our discussion by comparing something familiar, a protein, to a cell.
Continue reading