
Can’t live with them, can’t live without them. That just about sums up my relationship with Markov state models aka MSMs. They have known limitations, but they sure can be useful (sometimes) and also misleading (sometimes).
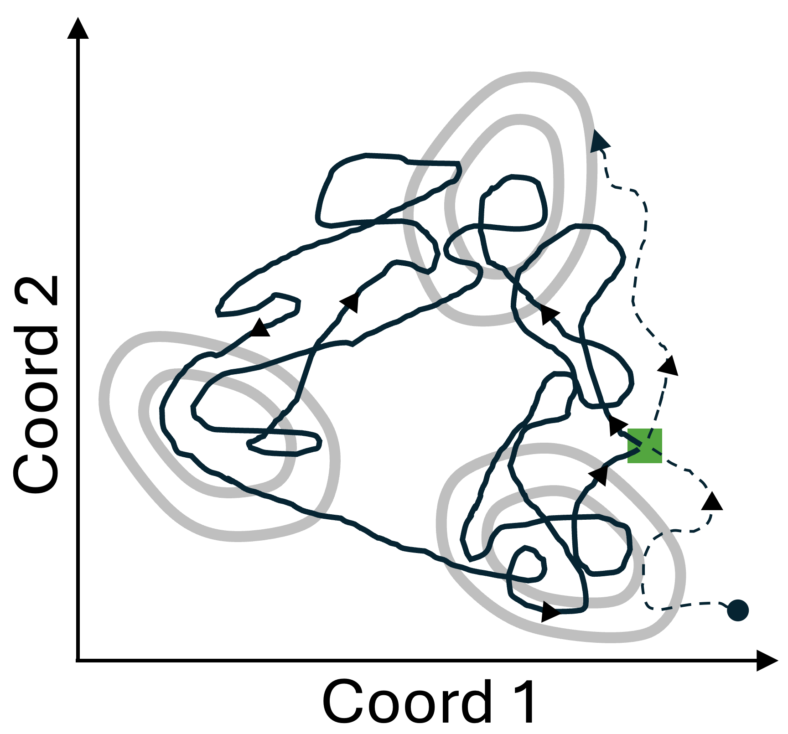
I wrote a few years ago about some of the limitations of MSMs, and the aim of this post is to expand on one point from that post, namely, the tendency of MSMs to simply recapitulate the distribution of the raw counts data when one or a small number of trajectories are used to train the model. That is, as first pointed out by Scalco and Caflisch, when analyzing a trajectory that revisits the same basins, you won’t learn anything from the MSM because it essentially spits back the quantities you would get from simple analysis, e.g., by counting populations.
Continue reading


 which we measure in a molecular dynamics (MD) simulation at successive configurations:
which we measure in a molecular dynamics (MD) simulation at successive configurations:  ,
,  ,
,  , and so on. Regardless of the length of our simulation, we can measure the average of all the values
, and so on. Regardless of the length of our simulation, we can measure the average of all the values  . We can also calculate the standard deviation σ of these values in the usual way as the square root of the variance. Both of these quantities will approach their “true” values (based on the simulation protocol) with enough sampling – with large enough
. We can also calculate the standard deviation σ of these values in the usual way as the square root of the variance. Both of these quantities will approach their “true” values (based on the simulation protocol) with enough sampling – with large enough  .
.


