

Count me among the weak of science.
Here I am again, feeling defensive, irate at reviewer critiques of our recent sub-Nobel prize work. Only in this case, the reviews are in my mind, yet to arrive. In fact, we haven’t even drafted the paper yet! But I can foresee what will happen. After all, if I’m honest, our contribution is clearly incremental.
Is it a failure? Were my expectations way off, again?
Continue reading


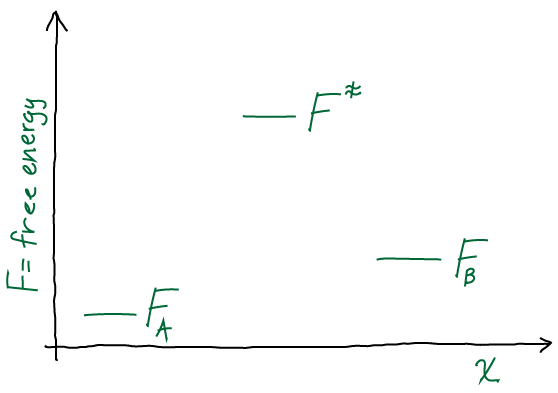
 ? But that was relaxation to equilibrium. Relaxation to a non-equilibrium steady state (NESS) is more interesting.
? But that was relaxation to equilibrium. Relaxation to a non-equilibrium steady state (NESS) is more interesting.

