

Such a beautiful thing, the PMF. The potential of mean force is a ‘free energy landscape’ – the energy-like-function whose Boltzmann factor exp[ -PMF(x) / kT ] gives the relative probability* for any coordinate (or coordinate set) x by integrating out (averaging over) all other coordinates. For example, x could be the angle between two domains in a protein or the distance of a ligand from a binding site.
The PMF’s basis in statistical mechanics is clear. When visualized, its basins and barriers cry out “Mechanism!’’ and kinetics are often inferred from the heights of these features.
Yet aside from the probability part of the preceding paragraph, the rest is largely speculative and subjective … and that’s assuming the PMF is well-sampled, which I highly doubt in most biomolecular cases of interest.
Let’s deal with each of the issues in turn: mechanism, kinetics, and sampling.
[* Note that a precise interpretation of the PMF requires knowledge of the Jacobian for the chosen coordinate(s): some intervals (x, x+dx) may contain more Cartesian configuration space than others.]Mechanism and the PMF
To my knowledge, David Chandler and coworkers were the first to highlight dangers of the PMF. They described energy landscapes – potential energy functions – with mechanisms that could not be described by obvious coordinate choices for PMF calculations (thus motivating path-sampling techniques). Expanding on those ideas, consider the two-dimensional landscapes below.



Although some of the landscapes perhaps could be described by some (tortuous) one-dimensional coordinate, in other instances that just wouldn’t be possible. Think how much worse the situation would have to be in a system like a protein with thousands of degrees of freedom.
Now take one more step on the road to skepticism and consider how PMFs tend to be constructed in computational studies. Because the calculations are so expensive, we tend to choose one or two coordinates in advance for study. These coordinate choices represent our pre-conceived ideas about how a system might function.
Based on the features of the PMF landscape, we build a story about mechanism … the dwells are here and the transition states there. And yet we know that all basins are not alike – some may be energy stabilized and other favored by entropy. The heights of barriers, their widths and likewise the dimensions of basins all are influenced by the particular coordinate(s) we chose in advance, as illustrated in the two-dimensional examples above.
So I would say the quantitative analysis of a PMF is inherently misleading, but the “stories” we build are almost more dangerous. Because our minds are naturally drawn to stories, such narratives are easy to retain. This wouldn’t be a problem if the stories were not biased by the subjective coordinate choices and perhaps also by inadequate sampling.
Is there a good way to think about mechanism? I believe the starting point has to be a trajectory ensemble of continuous unbiased transition events. Trajectories tell us the full sequence of events from an initial to a final state. And the ensemble reports on the diversity of mechanisms, something a PMF could never do. It’s true that trajectory ensembles are difficult to obtain, but at least they offer a potentially unbiased way to describe mechanism.
As a final perspective on mechanism, think about the goal of discovery. We use large amounts of computing resources and we would like to be able to discover something we did not know before. But if we only look at coordinates we presumed from the start to be important, we are severely impairing our ability to discover novel phenomena.
Perhaps it’s worth a moment to consider that our subjective coordinate choices typically are based on a further assumption – that we have good or complete knowledge of our system’s biological function. Is this really true?
Ideally, although it’s a challenge, we would perform unbiased analyses of path/trajectory ensembles to discover important coordinates. Significant work has already been done on automated discovery of coordinates, and I think such methodologies should play an increasingly important role in the future.
Does a PMF predict kinetics?
We’ve all done it – looked at a PMF (free energy landscape), estimated a barrier height, and tried to guess a rate constant for a process. If we haven’t tried to guess an absolute rate, we’ve at least said to ourselves something like, “Well, that barrier is higher than the other, so one process is slower than the other.”
But in principle, the PMF may not yield any information about kinetics at all! Look again at the two-dimensional landscapes above. Would you trust a one-dimensional PMF of any of these to give reliable rates? So why trust a projection from 1,000+ dimensions to one or two, as is usually done for biomolecular systems?
Again, I think part of the reason we over-interpret PMFs is because free energy landscapes speak to us like stories. We can’t resist the narrative that arises in the stat-mech part of our brains, even when we know better.
To drive home the point further, note that it’s even possible to construct a PMF that is exactly constant and yet does not exhibit diffusive dynamics. One way would be with a potential consisting of `anti-parallel’ valleys as in landscape (c) above, where the well depths and widths were tuned at each x value to exactly have equal probability when integrated over y.
Another constant-in-x PMF based on a non-trivial potential energy in x and y results from the following potential energy, which is a double-well potential in x modulated by a harmonic y component with x-varying width:
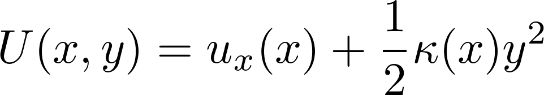
![]()
![]()
You can check this yields a constant PMF by integrating over y and multiplying the result by the Boltzmann factor of the energy minimum at any x value. Below is a sample trajectory of x values (y not shown) simulated with overdamped Langevin (a.k.a. Brownian) dynamics.

Clearly the behavior is not simple diffusion in x, even though the PMF(x) is constant. (Histogramming a lot of these trajectories into x bins numerically confirms the PMF is flat.) And that’s no surprise, from a theory standpoint. The PMF generally does not govern dynamics, except in rare cases where an ideal reaction coordinate has been used … and suitable effective dynamics have been specifically derived for the selected coordinate.
Are biomolecular PMFs well sampled?
Very sophisticated simulation and analysis techniques are used to calculate PMFs. But should we trust the results? To be clear, I’m not questioning the rigor behind the methods, but rather the likelihood that the PMFs are well-sampled.
Consider the popular analysis approach WHAM (weighted histogram analysis method). WHAM seeks to provide the best possible PMF given the data available from the different simulation windows. This is a very different goal from assessing whether sufficient sampling has been performed. For better and for worse, WHAM almost always provides a smooth estimate of the free energy profile. Our minds tend to confuse smoothness with reliable, well-sampled data. But the two things are completely independent.
Let’s be more concrete. How many nsec are typically used in a single window of a WHAM protein calculation? Often the answer seems to be 10 nsec or less. Is that really enough time to sample even a constrained protein process? When careful studies are done on small peptides, they suggest 10s of nsec are needed for good sampling of these tiny systems!
Now consider a thought experiment. If indeed every window of a WHAM-like PMF calculation is well sampled, then we have an equilibrium ensemble for that window. And if the PMF is accurate, we also know the relative free energy of that coordinate value. We therefore can generate an overall equilibrium ensemble by combining all the window ensembles, weighting each by the Boltzmann factor of the window-specific PMF. Would you trust this equilibrium ensemble? Further, an equilibrium ensemble can be projected onto any coordinate to generate a new PMF. Would you trust that new PMF?
Pessimism
To sum up: Be careful! I am dubious that protein PMFs are well-sampled. But even when a PMF is exactly accurate, it reveals a landscape based on a subjectively chosen coordinate set. Physical scientists are adept at building a story from a landscape, but any given landscape is likely to be deceptive both in terms of mechanism and kinetics. Perhaps it’s time for us to move on from over-reliance on the PMF.
Acknowledgement.
I very much appreciate comments on a draft given by Alan Grossfield.
Further reading
Dellago, C.; Bolhuis, P. G.; Csajka, F. S. & Chandler, D., Transition path sampling and the calculation of rate constants, J. Chem. Phys., 1998, 108, 1964-1977
Grossfield, A. & Zuckerman, D. M., Quantifying uncertainty and sampling quality in biomolecular simulations, Annu Rep Comput Chem, 2009, 5, 23-48.
Kumar, S.; Rosenberg, J. M.; Bouzida, D.; Swendsen, R. H. & Kollman, P. A., Multidimensional free-energy calculations using the weighted histogram analysis method, J. Comput. Chem., 1995, 16, 1339-135
Lyman, E. & Zuckerman, D. M., On the Structural Convergence of Biomolecular Simulations by Determination of the Effective Sample Size, J. Phys. Chem. B, 2007, 111, 12876-12882
McGibbon, Robert T., Brooke E. Husic, and Vijay S. Pande, Identification of simple reaction coordinates from complex dynamics, The Journal of Chemical Physics 146, 044109 (2017)
Zuckerman, D.M., Statistical Physics of Biomolecules: An Introduction, CRC Press, 2010.